Background and Overview
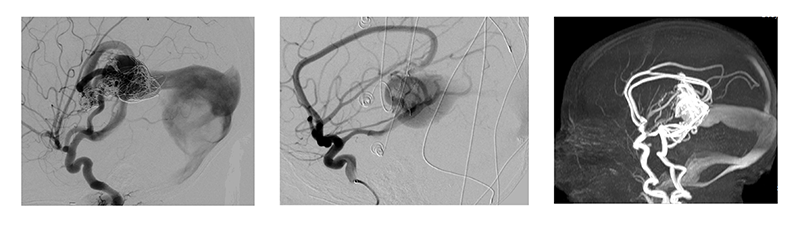
Vein of Galen malformation (VOGM) is the most severe vascular anomaly affecting the pediatric brain. Shunting between choroidal arteries and the embryologic prosencephalic vein may cause congestive heart failure, hydrocephalus, epilepsy, and impaired brain development. Fundamental gaps in our understanding of the genetic and molecular basis of VOGM impede therapeutic development. To address these obstacles, we will elucidate the genetic basis of VOGM through identification of germline and somatic mutations through in situ analysis of the VOGM lesion enabled by endoluminal tissue sampling (ETS). To acquire a critical mass of genetic information, we establish the VOGM Genetics Research Consortium (VOGM-GRC).
Our central hypothesis is that inadequate access to VOGM tissue in situ hinders the identification of somatic mutations responsible for VOGM pathogenesis and maintenance. While prior whole exome sequencing (WES) studies of VOGM identified a genome-wide significant burden of rare, damaging mutations in EPHB4, most cases remain genetically unsolved. To overcome this limitation, we have successfully used ETS to safely collect ~600 endothelial cells from 5 individual VOGM lesions during standard clinical care. In addition to screening for novel somatic mutations, we will further define the genomic landscape through transcriptional analysis, i.e., single-cell RNA sequencing of cells obtained from ETS.
VOGM is the most common and severe neonatal cerebrovascular arteriovenous malformation (AVM). Aberrant connection between primitive choroidal arteries and the median prosencephalic vein of Markowski leads to intracranial hypertension, congestive heart failure and hydrocephalus. Endovascular embolization is the first line treatment due to improved mortality and neurological outcomes. Nevertheless, many VOGMs remain under-treated or experience complications from embolization, especially when performed during the first year of life. The prognosis for VOGM varies depending on the severity of the malformation and the level of treatment received. With early diagnosis and appropriate treatment, the long-term outlook can be good, particularly in cases where the malformation is not extensive and can be effectively managed with medications, surgery, and rehabilitative therapy. However, in severe cases, the prognosis can be poor, as the abnormal pressure on the brain can cause irreversible damage and can even be fatal in some cases.
Our limited understanding of the molecular, genetic, and pathophysiological basis of VOGM has impeded development of non-invasive pharmacotherapies. Most VOGM are considered sporadic, isolated congenital lesions, but Mendelian disorders also display VOGM as a pathologic feature (1). Recent evidence suggests that germline mutations in chromatin modifier and Ephrin genes account for ~15% of cases (2), however the majority of cases are genetically unsolved. Identifying additional genetic drivers and mechanisms underlying VOGM will improve our understanding of the pathophysiologic basis of the disease and may inform pharmacological or gene-based treatment strategies.